Network Inference
Current Participants: Nan Papili Gao
The inference of biological networks has important applications, from finding treatment of diseases to engineering of microbes to produce drugs and biofuels.
Biological networks are often drawn as a graph, in which nodes represent biological entities and edges indicate biological connections. These connections are used to describe a variety of biological functions, for example physiochemical interactions (e.g. binding between molecules), chemical transformation (from substrate to product), regulation (activation/inhibition), gene transcription, protein translation, and many more. Inferring such networks from data means identifying the interconnections (or the edges) among biological entities (or the nodes). A subset of this problem that is of particular interest in this project, is the identification of directed edges or arrows. This directionality can be generally thought to mean causality, i.e. A --> B means “A causes B”. The inference of such causal networks from (time-series) biological data is difficult and has motivated community-wide challenges for the creation and assessment of inference methods (see Dialogue on Reverse Engineering Assessment and Method or external page DREAM challenge). Despite tremendous efforts from different disciplines to tackle this problem, resulting in hundreds of algorithms and examples, such inference is still very much an unsolved problem.
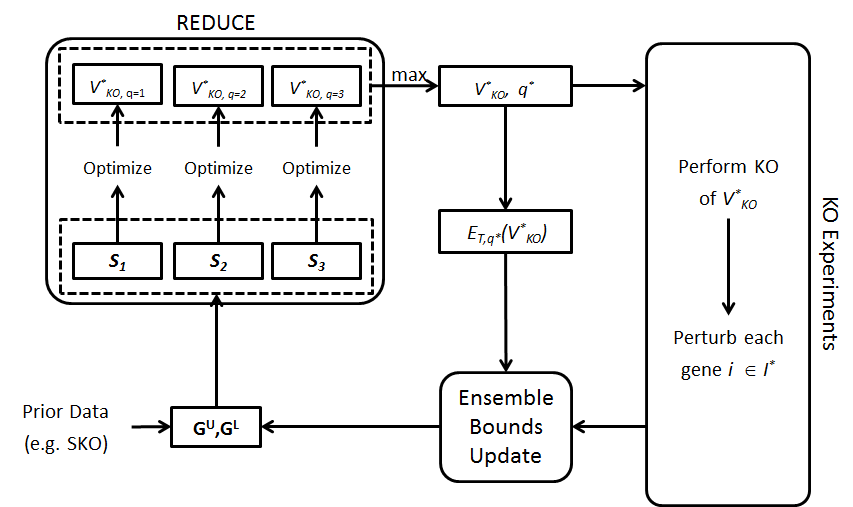
The overall goal of this project is to give answer to the challenge of biological network inference, particularly gene regulatory networks (GRNs), using a two-pronged approach. First, we have developed theoretical framework and algorithms for ensemble inference, called TRaCE, using which we could produce an ensemble of networks that are consistent to and therefore indistinguishable by the available data. This ensemble thus represents the uncertainty in the network inference, and provides a direct measure of network inferability. This uncertainty is a consequence of the lack of information in the data to uniquely determine the network structure, for example because of suboptimal experimental design. For this reason, complementing the ensemble inference, we have created an optimal design of gene knock-out experiments for GRN inference, called REDUCE. In several examples, the iterations of TRaCE and REDUCE could fully resolve the GRN inference problem (see Figure 1), producing unique network structures while employing a small set of gene KO data.
References
- Papili-Gao, N., Ud-Dean, SMM. and Gunawan, R. SINCERITIES: Inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles, 2016. (submitted to Bioinformatics, available in external page biorxiv)
- Papili-Gao, N., Ud-Dean, SMM. and Gunawan, R. Gene regulatory network inference using time-stamped cross-sectional single cell expression data. IFAC-PapersOnLine 49(26): 147-152. external page abstract
- Ud-Dean, SMM., Heise, S., Klamt, S. and Gunawan, R. TRaCE+: Ensemble inference of gene regulatory networks from transcriptional expression profiles of gene knock-out experiments. BMC Bioinformatics 17, 252 (2016). external page abstract
- Ud-Dean, SMM., and Gunawan, R. Optimal design of gene knock-out experiments for gene regulatory network inference. Bioinformatics (2015). external page abstract
- Ud-Dean, SMM., and Gunawan, R. Ensemble Inference and Inferability of Gene Regulatory Networks. PLoS ONE, 9(8), e103812 (2014). external page abstract